Home → autoinflamatorias y autoinmunes
Psoriasis, Dermatitis Atópica, Hidradenitis Supurativa, Pioderma Gangrenoso, Pénfigo/Penfigoide, Vitiligo, Dermatitis Herpetiforme/Enfermedad Celíaca, Lupus Eritematoso.
En los últimos años hemos sido participes de una revolución en cuestión de descubrimientos en el campo de la fisiopatogenia y, como consecuencia de ello, el advenimiento de una catarata de novedades e innovaciones en el ámbito terapéutico en un grupo de procesos, hasta ayer, dermatológicos, y hoy considerados sistémicos, ayer, psicosomáticos y hoy, autoinmunes y autoinflamatorios, ayer, sin opciones terapéuticas y hoy, invadidos por un aluvión de nuevas opciones.
En este grupo de enfermedades encontramos algunas muy conocidas y temidas, como Psoriasis, Dermatitis Atópica y Vitiligo y, otras, desconocidas, aún para la población médica, como Hidradenitis Supurativa, Pioderma Gangrenoso o Pénfigo/Penfigoide. Aquí dedicamos unas líneas a las reconocidas, pero en lo sucesivo iremos sumando otros procesos menos reconocidos.
Nuestra intención es darle conocimiento y posibilidades de atención a quien es portador de estas enfermedades y a los profesionales que desconocen cómo enfrentar estos desafíos. Pretendemos brindar apoyo a aquel Profesional de la Salud que lo necesite y evacuar toda duda surgida.
PSORIASIS.Se estima que 100 millones de personas en todo el mundo (2 a 3 de cada 100) conviven hoy con el problema de la psoriasis, viendo seriamente afectada su vida cotidiana. Se estima que entre el 10 y el 30 % de estos pacientes desarrollarán artritis psoriásica.
Existe un gran desconocimiento a escala social y una información limitada de lo que es la psoriasis, lo que lleva a situaciones de ansiedad y temor al ser diagnosticada.
La psoriasis es una enfermedad crónica de la piel, no contagiosa, que se caracteriza por una actividad inflamatoria anormal, que se asocia al crecimiento excesivo de los queratinocitos (un tipo de células de la piel) y que puede cursar, además, con afectación de otros sitios del organismo, como por ejemplo las articulaciones.
Se manifiesta con evidentes signos físicos como eritema (enrojecimiento) y lesiones escamosas, resecas, engrosadas e inflamadas de la piel, variando la extensión de piel afectada de unos individuos a otros. Estas lesiones pueden resultar muy molestas para el enfermo que padece psoriasis, manifestándose con prurito (picazón), sensación urente o con dificultades en la movilidad, entre otros síntomas.
La psoriasis afecta aproximadamente 2 a 3 % de la población mundial (entre 80 y 100 millones de personas en todo el mundo). En Argentina, existe alrededor de un millón de afectados, de los que el 70 % la padece de modo leve, sufriendo un 20 % su variante moderada o grave. Aproximadamente un 15 % de los pacientes presenta espontáneamente remisiones prolongadas, mientras que otro tanto cursa de modo intermitente con etapas de mejoría y empeoramiento alternadas.
Suele aparecer entre los 15 y los 35 años, aunque se puede presentar a cualquier edad. De hecho, entre el 10 y el 15% de quienes la padecen, la manifiestan antes de los 10 años. Afecta por igual a personas de ambos sexos.
La psoriasis requiere un tratamiento y un control continuo, alternando y combinando en muchas ocasiones varias opciones terapéuticas.
Además de los signos externos, la psoriasis tiene un importante impacto sobre la calidad de vida de los pacientes, por cuanto afecta muy seriamente al desarrollo de las actividades cotidianas y posee un marcado componente de afectación psicológica: cuadros depresivos, aislamiento, rechazo estético, social y laboral, vergüenza, etc. De hecho, su impacto sobre la calidad de vida del enfermo puede compararse, o incluso superar, al de otras enfermedades crónicas como la hipertensión, el cáncer o las enfermedades coronarias.
El mejor conocimiento, por parte de la sociedad, de lo que significa psoriasis y de lo que supone para el psoriásico es, sin duda, el primer paso hacia la plena integración del enfermo. Ello requiere la colaboración y el compromiso de todos los implicados, enfermos, familiares y allegados a los enfermos, profesionales de la medicina, administradores de los sistemas de salud, organizaciones gubernamentales y ONG, entre otros. De este compromiso surgirán las ideas y caminos que llevarán a una mejor calidad de vida a los que padecen esta enfermedad.
¿Qué es la psoriasis?
Es una enfermedad inflamatoria, crónica, no contagiosa. Se presenta en forma de lesiones rojas, cubiertas por escamas blancas, secas, que se localizan en codos, rodillas, tronco y cuero cabelludo. Puede comprometer las palmas de las manos y las plantas de los pies, las uñas, la semimucosa labial y la mucosa genital.
¿Es contagiosa la psoriasis?
No, no se puede contraer psoriasis por el contacto con otras personas.
¿A quiénes afecta?
La psoriasis afecta entre el 2% y 3% de la población de la Argentina. Puede aparecer en hombres o mujeres indistintamente. Puede presentarse a cualquier la edad, tanto en lactantes, niños y adultos.
¿Cuál es la causa de esta enfermedad?
Está relacionada con el sistema inmunológico, existe una predisposición genética y múltiples factores ambientales que desencadenan o agravan la enfermedad.
Los factores infecciosos (bacterianos, virales y hongos), determinados medicamentos, el consumo de alcohol y tabaco, el stress, los climas fríos, el rascado o fricción frecuente, cambios hormonales, pueden ser los disparadores de la enfermedad y/o empeorar el estado del enfermo. Las lesiones de la piel se ven descamativas, dado que el proceso normal de recambio de las células de la capa superficial de la piel es de 28 días y en los pacientes con psoriasis este proceso está acelerado y lleva entre 3 y 4 días. A esto se suma la inflamación, que se manifiesta con enrojecimiento y engrosamiento de las lesiones.
¿Cuáles son los diferentes tipos de psoriasis?
1. Psoriasis vulgar: es la más frecuente. Son placas rojas cubiertas por escamas blancas, secas, que se desprenden fácilmente. Pueden tener distintos tamaños y formas.
2. Psoriasis en gotas: corresponde a pequeñas lesiones rojas, milimétricas con escamas blanquecinas. Son múltiples, de aparición brusca, generalmente en niños y adolescentes, posteriores a un proceso infeccioso de vías aéreas superiores y se ubican en el tronco y extremidades.
3. Psoriasis invertida: A diferencia de la vulgar son lesiones sin escamas, localizadas en los pliegues de la piel como axilas, ingles o debajo de los mamas. Pueden fisurarse.
4. Psoriasis pustulosa: Generalizada: es una forma clínica severa, infrecuente, con compromiso del estado general del paciente y se aconseja su internación. La psoriasis pustulosa puede ser localizada a nivel de las palmas de las manos o en las plantas de los pies.
5. Psoriasis eritrodérmica: es un tipo infrecuente, muy severo y también requiere internación. Generalmente es secundario a tratamientos incorrectos o infecciones y se presenta con una coloración roja generalizada que compromete entre el 80% y la totalidad de la superficie corporal. Suelen presentar fiebre, escalofríos, y alteraciones hemodinámicas.
6. Psoriasis ungueal: existe un porcentaje importante de compromiso de las uñas con hoyuelos, destrucción de parte de la uña o con material debajo de las mismas. En la artropatía psoriásica el compromiso de las uñas suele ser importante.
7. Psoriasis artropática o artritis psoriásica: afecta principalmente articulaciones periféricas y también puede afectar la columna vertebral.
8. Psoriasis del pañal: cuando la psoriasis afecta a niños pequeños que aún utilizan pañal, las lesiones pueden aparecen preferencialmente en la zona cubierta por el mismo, como respuesta a la irritación.
¿Puede una persona padecer psoriasis sin saberlo?
Efectivamente algunas manifestaciones de la psoriasis pasan ocasionalmente inadvertidas. Especialmente las afectaciones de las uñas pueden ser interpretadas erróneamente como micosis y tratadas por gente no especializada con anti-micóticos. Esto a veces es de gran importancia dado que algunos de estos enfermos desarrollarán artritis psoriásica y su diagnóstico sufrirá un retraso sumamente riesgoso. Las lesiones de la piel, cabello y uñas deben ser siempre consultadas con el dermatólogo.
¿Qué es la artritis psoriásica?
Es una forma clínica de psoriasis que afecta las articulaciones. Es un tipo de artritis que provoca inflamación e hinchazón en manos, pies o articulaciones como las rodillas, caderas, codos y columna. El 70% de los pacientes presentan primero una psoriasis cutánea y luego articular, el 15% se presenta en forma conjunta y un 15% es solo articular.
¿Qué riesgo tiene un paciente con psoriasis de padecer artritis psoriásica?
Aproximadamente entre el 10 % al 30% de las personas que padecen psoriasis pueden desarrollar psoriasis artropática. La afección cutánea en la piel precede a la artritis en 10 años.
¿Qué características particulares tiene la artritis psoriásica?
La artritis psoriásica es un tipo específico de artritis que causa dolor, rigidez e inflamación articular. Puede aparecer en manos, pies, tobillos y otras articulaciones. Su diagnóstico temprano y tratamiento pueden mejorar el dolor y la inflamación retrasando y evitando el daño progresivo de las articulaciones. Sin tratamiento la enfermedad puede potencialmente derivar en discapacidad.
¿Con qué síntomas se puede presentar la artritis psoriásica?
Dolor e inflamación de las articulaciones.
Rigidez y dolor matinal.
Inflamación que hace ver los dedos de las manos y pies como si fueran salchichas.
Alteraciones a nivel de las uñas.
Fatiga generalizada.
¿En qué formas clínicas puede presentarse la artritis psoriásica?
1. Asimétrica: puede involucrar pocas o varias articulaciones pero no en el mismo lado del cuerpo.
2. Simétrica: es parecida a la artritis reumatoidea. Afecta usualmente articulaciones pares y simétricas en el cuerpo.
3. Interfalángica distal: afecta primariamente los dedos de la mano y pies, en la parte cercana a las uñas.
4. Espondilitis: es una inflamación de la columna vertebral que suele afectar a pacientes con artritis psoriásica. La inflamación está acompañada con rigidez de cuello, cintura.
5. Artritis mutilante: es una artritis severa, deformante y destructiva.
6. Entesitis: inflamación con dolor a nivel de la inserción de uno o varios tendones.
¿Cuáles son los tratamientos posibles?
Para la psoriasis cutánea existen múltiples tratamientos según el tipo de psoriasis: tratamiento locales: lociones, cremas o ungüentos, suelen utilizarse en psoriasis leve y como complemento de otros tratamientos. En formas clínicas moderadas o severas se indica fototerapia o tratamientos sistémicos, en forma combinada, rotativa o intermitente. En el tratamiento de la artritis psoriásica se recomienda la utilización de terapias que mejoren los síntomas relacionados con el dolor y la inflamación articular (anti-inflamatorios), que modifiquen el curso de la enfermedad (metotrexato, etc) y mejoren la capacidad funcional de los pacientes (programa de ejercicios, terapia física, etc). En determinados pacientes se pueden utilizar, según la severidad del cuadro, los sitios comprometidos y la respuesta a otros tratamientos, distintos agentes biológicos como el etarnecept, el infliximab, el adalimumab y el efalizumab, entre otros.
La visita al médico especialista es indispensable para determinar que tipo de tratamiento es el correcto a seguir.
¿Quién trata la psoriasis y la artritis psoriasica?
Si el compromiso de la enfermedad es sólo cutáneo, los dermatólogos serán los médicos tratantes y si el compromiso es sólo articular, los reumatólogos. En los casos con daño conjunto cutáneo y articular, el tratamiento en equipo entre el dermatólogo y el reumatólogo es el indicado.
Pida ayuda, porque sino nadie va a saber que usted la necesita.
Cuanta más gente conozca y comprenda la psoriasis, más fácil y mejor va a ser para usted.
Acepte hablar de su psoriasis con otros, al punto de sentirse cómodo.
Sus verdaderos amigos van a querer saber cómo está y cómo lo pueden ayudar. No se van a alejar porque tenga psoriasis.
No hay nada de que avergonzarse o sentirse incómodo.
Una enfermedad de la piel no es más que eso, una enfermedad. Aún cuando algunas personas le atribuyan cosas raras.
Es natural que se sienta ansioso, enojado o deprimido. Los amigos pueden ayudar. La gente a su alrededor puede ser un buen soporte. Puede ayudar a sus amigos haciéndoles saber que la psoriasis no es contagiosa y que es el resultado de que las células epidérmicas se reproduzcan rápidamente.
La consulta en centros especializados le aportará alternativas para el control de sus lesiones cutáneas y/o articulares, le será de gran ayuda para terminar de comprender su enfermedad y le dará contención emocional.
IPSO es un centro especializado en el diagnóstico y tratamiento de la psoriasis. Cuenta con un equipo interdisciplinario capaz de ofrecerle las alternativas terapéuticas actuales en psoriasis. Este incluye a médicos dermatólogos, reumatólogo, clínica médica y psicóloga con experiencia en el manejo de esta patología.
En IPSO contamos con fototerapia ultravioleta, la cual ayuda a un gran número de enfermos de psoriasis, produciendo en ellos remisión de las lesiones por un tiempo prolongado luego de haber completado un ciclo de tratamiento.
Los médicos de IPSO tenemos experiencia en emplear todo tipo de medicaciones para el tratamiento de la psoriasis, incluyendo a los biológicos, por lo que podemos hacer un uso racional y efectivo de las mismas.
En IPSO le dedicamos tiempo a la consulta del enfermo de psoriasis. Sabemos que muchas veces la problemática de esta enfermedad no puede ser resuelta en los breves tiempos que se suelen establecer para las consultas, y por ello hemos duplicado o triplicado según el caso dichos lapsos. Contemplamos también la demanda espontánea, sin turno previo, dado que por sus características el paciente con esta enfermedad frecuentemente requiere de este servicio.
Finalmente, en IPSO encontrará comprensión y contención. Conocemos la psoriasis, sabemos que impacta profundamente en la vida de quienes la padecen y tenemos siempre la mano tendida para recorrer el camino juntos, hacia el control sostenido de esta enfermedad.
¿Qué es la Dermatitis Atópica?
Dermatitis Atópica (DA) es una enfermedad cutánea, crónica, inflamatoria, caracterizada por prurito, sequedad y liquenificación, que históricamente prevaleció en la infancia, pero en la actualidad ha aumentado su presencia en la edad adulta, bien por persistencia desde la infancia o con su aparición ya en la adultez.
Se considera una enfermedad crónica, persistente, no maligna, pero sin poner en riesgo la vida del paciente, altera significativamente su calidad de vida. Sus manifestaciones cutáneas aumentan el riesgo de infecciones, alergias y patologías psiquiátricas. Su estudio profundo ha determinado su carácter multisistémico y sus implicancias socioeconómicas (mayor gasto en salud y menor productividad por dificultades en la vida diaria).
Definición:
Enfermedad benigna, crónica, recurrente, no infecto-contagiosa, inflamatoria y pruriginosa de la piel, que afecta a cualquier edad, niños y adultos, pero predominantemente a los primeros, con una distribución corporal típica de acuerdo a la edad. Se asocia a niveles elevados de IgE, historia personal o familiar de alergias (rinitis, asma, alimentaria). Algunos pacientes pueden cursar con niveles de IgE normales y con ausencia de estos tipos de alergia.
Definición de Enfermedad Moderada a Severa.
Para considerar DA moderada a severa debemos contar con los siguientes criterios:
Compromiso mayor al 10% de la superficie cutánea,
Lesiones con características moderadas a severas,
Compromiso de áreas altamente dificultosas por su visibilidad o funcionalidad, por ejemplo cara, manos, pies, cuello,
Impacto importante sobre la calidad de vida (prurito, sueño, actividad laboral),
Scores: SCORAD mayor a 25, EASI mayor a 7.
¿Cómo se llega al Diagnóstico de Dermatitis Atópica?
El diagnóstico actual de DA se alcanza por reunión de criterios. Aquí la biopsia de piel no tiene relevancia debido a la ausencia de signos precisos.
Estos se clasifican en mayores y menores, en función de su importancia.
Criterios Mayores.
Prurito
Características y distribución de las lesiones
Historia personal o familiar de atopía
Dermatitis crónica y recidivante
Criterios Menores.
Xerosis, ictiosis, hiperlinearidad palmar, queratosis pilar, IgE elevada, edad de comienzo temprana, tendencia a infecciones cutáneas, dermatitis inespecífica de manos y pies, eccema de pezón, queilitis, conjuntivitis recurrente, pliegue infraorbitario, queratocono, catarata su cápsulas posterior, oscurecimiento palpebral, eritema/palidez facial, pitiriasis alba, pliegues en cara anterior de cuello, prurito con la sudoración, intolerancia a lana y solventes, acentuación perifolicular, intolerancia a alimentos, etc.
En otro orden de cosas, podemos definir cuáles de estos son los más importantes definiéndolos como características esenciales, características importantes y características asociadas: Podemos incluir en las esenciales al prurito, dermatitis eccematosa (aguda, subaguda y crónica) y la evolución crónica y por brotes; y dentro de las segundas, las importantes, debemos mencionar a edad temprana de inicio, atopía, antecedentes familiares de atopía, aumento de IgE total, xerosis y liquenificación. Finalmente, entre las asociadas encontramos: queratosis pilar, pitiriasis alba, hiperlinearidad palmar, ictiosis, trastornos oculares y palpebrales, eccema de pezón y periauriculares, hiperqueratosis folicular, liquen simple crónico, prurigo nodular, eccema numular y eritrodermia.
Comorbilidades
Conocemos como Comorbilidad a todo aquel proceso que tiene fuerte relación de asociación con la patología de base. En DA los principales procesos considerados en este apartado son gastrointestinales (enfermedad inflamatoria intestinal), dermatológicos (alopecia areata, vitiligo, urticaria crónica espontánea) y reumatológicos (artritis rematoidea).
Aspectos Cotidianos.
Los expertos explican que la dermatitis atópica es una afección cutánea crónica, recurrente y generalizada que comienza en la infancia y continúa en la edad adulta en la mayoría de las personas. Las personas con dermatitis atópica grave pueden necesitar hospitalización para recibir tratamiento. Los comentarios de las organizaciones de pacientes y profesionales destacan que la condición es debilitante y aislante, que afecta todos los aspectos de la vida (física, psicológica, social y financiera). Remarcan que, si la condición es severa, se asocia con picazón intolerable que interrumpe el sueño, altera la vida laboral y social, y existe un mayor riesgo de depresión y suicidio.
¿Qué significa Fenotipo y Endotipo? ¿Para qué sirve?
Fenotipo se define como el grupo de características que define a esta enfermedad y sin importar los mecanismos que la producen; mientras que endotipo interesa a los mecanismos moleculares de producción de la enfermedad. Es una alternativa de clasificación de la enfermedad según mecanismos de producción, edad o raza.
Biomarcadores son indicadores medibles a cerca de alguna característica de la enfermedad, como diagnóstico, pronóstico, o respuesta al tratamiento.
Las variantes fenotípicas de la DA son: DA del niño, DA del adulto, DA intrínseca y DA extrínseca. La DA de inicio en la Infancia, en la forma clásica de la enfermedad y la más frecuente históricamente, mientras la DA del Adulto, es la variante más reciente y que frecuentemente conlleva al error diagnóstico terapéutico. El 15% de los niños padecen DA y muchos de ellos persisten con esa condición en la vida adulta. En el niño predominan manifestaciones agudas y subagudas, eccematosas, exudativas, en zonas de extensión, en el lactante; afectando grandes pliegues en la infancia temprana.
La DA del Adulto presenta 3 variantes clínicas, siendo la más frecuente (20-30%) DA del Adulto Persistente, cuando el paciente lo es desde la infancia, afectando claramente los grandes pliegues, la cara y variablemente, el cuerpo y las manos; DA del Adulto Recidivante, de este grupo participan pacientes que siendo atópicos desde la infancia, desarrollan una evolución clásica de DA, resolviendo su enfermedad a inicios de la adolescencia, pero que inesperadamente, reincide en la edad adulta. De difícil diagnóstico, la variedad más frecuente es el eccema crónico de manos, asociada a actividades laborales y domésticas y, por último, DA de Inicio en la Adultez, observada más frecuente entre 20-40 años y, menos frecuentemente, en personas más añosas, con patrones clínicos muy infrecuentes, como eccema numular, prurigos o distribución en cabeza y cuello.
A que denominamos DA Extrínseca e Intrínseca.
Ambas son formas de presentanción de la enfermedad, donde la primera corresponde al 80% de los casos, y se caracteriza por IgE elevada, antecedentes familiares de DA e IgE específica para alértenos alimentarios y aéreos. En tanto, la variante Intrínseca no presenta estas características. La eosinofilia se encuentra con mayor presencia en la variante extrínseca.
Mecanismos de Producción.
La Dermatitis Atópica es una enfermedad que, inmunologicamente, se encuentra mediada por la célula T y, principalmente, de tipo Th2. Los distintos aspectos de la enfermedad están dados por distintas vías inmunológicas. Una de ellas es común a todos los aspectos, la Th2 (IL4-IL5-IL13). Otras dos vías son responsables de la evolución de la enfermedad, variable según el Fenotipo. Estas son Th22/IL22 y Th17/IL23. Las dos primeras, Th2 y Th22, se encuentran presentes en las lesiones agudas, con defecto en la finalización de la diferenciación del queratinocito, dando por resultado una alteración de la barrera cutánea.
El complejo de diferenciación epidérmico está determinado por genes, que pueden verse afectado por disminución de su expresión por acción de IL4 y, por lo tanto de la barrera cutánea, por defecto en la síntesis de filigrina, loricrina e involucrina. En asociación con IL13, esta IL4, afecta la expresión del gen de la filagrina, aún sin estar presente la mutación de esta proteína. Aquellos pacientes donde la mutación en el gen de la filagrina, manifiestan su enfermedad de forma más severa y persistente, pero sólo representa el 30% del total de los pacientes con DA.
Tratamiento.
Clásicamente las alternativas de tratamiento para DA son: corticosteroides tópicos como primera línea de tratamiento, inhibidores de la calcineurina como segunda línea, en tercera línea de tratamiento, fototerapia y, en la cuarta línea de ataque, los inmunosupresores sistémicos. En esta última encontramos a Ciclosporina (única autorizada por prospecto), metotrexato, azatioprina y micofenolato mofetil. Estos fármacos requieren múltiples controles de laboratorio y presentan eventos adversos importantes. Si uno de estos no es efectivo por largos períodos de tratamiento, y recae en breve, debe ser sustituido por otro.
Todo tratamiento debe ser comenzado y suplementado con educación, soporte psicológico, emolientes e hidratantes, vendajes y hospitalización.
Los rebrotes severos deben ser contenidos con corticosteroides de alta potencia o sistémicos por cortos períodos de tiempo.
Dupilumab.
Se considera la quinta línea de tratamiento. Esta alternativa debe ser propuesta cuando al menos haya fracasado a un inmunosupresor sistémico. Es un anticuerpo monoclonal dirigido a bloquear la subunidad alfa del receptor de la IL 4, bloqueando la señalización tanto de esta como de la IL 3.
Este medicamento no se indica en cualquier ocasión en un paciente portador de DA severa, aunque posea los criterios de gravedad, para ello debe no poder controlarse la enfermedad con todas las medidas terapéuticas antes descriptas o en el caso de que aquellas estén contraindicadas. En aquellos casos donde es indicado y administrado, Dupilumab es muy eficaz y seguro, con muy rápido inicio de acción.
Entre los efectos adversos, la conjuntivitis es el evento adverso más frecuente (37-53%). Pero también se han reportado blefaritis, queratitis, herpes simple, enrojecimiento facial, alopecia y artralgia.
Upadacitinib.
Es un nuevo fármaco de investigación ya aprobado para su uso en Estados Unidos por FDA (Agencia de Alimentos y Medicamentos), inhibidor de Janus Kinasa (JAK) de administración oral, con gran poder en la inhibición de JAK1, JAK2, JAK3 y Tirosin Kinasa2. Utiliza la vía de la IL31 mediadora del prurito, esta citokina está involucrada en la señalización de DA a través de JAK. Ya fue aprobado en varios países para su uso en artritis reumatoide, muy eficaz en esta así como en artritis psoriásica y espondilitis anquilosante. Entre los eventos adversos descriptos se observó, al igual que en AR, erupción acneica, aunque de todas maneras presenta, luego de finalizados los estudios fase III, un excelente perfil de seguridad. En conjunto, las Ils 4-13-31, son conocidas citokinas pruritogénicas, conductores de la inflamación cutánea en la DA y requieren, en un punto de la cascada, de JAK1 para su acción biológica, conduciendo la señalización del prurito crónico.
Baricitinib.
Posicionado como quinta línea de tratamiento, junto con dupilumab. Indicado en tratamiento de la enfermedad moderada a severa e indicado luego de haber fallado a, al menos, un inmunosupresor sistémico. En los estudios en curso asociado a corticosteroides tópicos vs placebo, muestra mejoría del prurito y de calidad de vida, evaluado por EASI 50-75 y por DLQI 0-1 a la semana 16 de tratamiento.
Generalidades.
Es una enfermedad crónica, inflamatoria, recurrente e invalidante, que genera una carga importante al paciente y está asociado a desórdenes variados, lo que reduce significativamente la calidad de vida (QoL), como depresión, estigmatización, inactividad, alteraciones en el orden laboral, dificultades en la vida sexual y varios factores de riesgo cardiovascular. Su origen es multifactorial.
El 1º simposio sobre HS, se desarrolló en Dessau, (2006), y formuló la siguiente definición: Es una enfermedad crónica, inflamatoria, recurrente e invalidante del folículo del pelo terminal, que usualmente se presenta luego de la pubertad, dolorosa, ubicado profundamente, con lesiones inflamatorias en la glándula apócrina, en las zonas de pliegues, como axilas, ingles y región anogenital.
Epidemiología.
La prevalencia reportada en anteriores estudios son de un amplio rango, de 0.4-4%. Un estudio reciente una prevalencia de 1%, en una muestra representativa de la población francesa. En un estudio danés, basados en datos prospectivos, la prevalencia fue superior a 4.1%, y en la examinación luego de un año, fue de 1% (intervalo de confianza 0.4-2%).
La mujer es más frecuentemente afectada, la relación H:M es 1:2-1:3.3. Una excepción a esto es el compromiso perianal, donde los hombres predominan. En USA la prevalencia es significativamente menor (0.05%). Algunos datos de incidencia en USA hallan 6/100000 hab en 2008.
Rara vez la HS se desarrolla antes de la pubertad y luego de la menopausia, sin embargo, la persistencia de las lesiones luego de la menopausia no es inusual. El reporte dice que sólo el 2% ocurre antes de los 11 años, sin embargo el corte de estudios indican un inicio prepuberal temprano en el 7.7%. El rango de edades de inicio es de 23 años.
Fisiopatogenia.
El mecanismo de producción de la enfermedad está centrada en la unidad pilosebácea apócrina, su inflamación y su taponamiento por hiperqueratosis folicular, son el evento primario de desarrollo de la enfermedad en individuos genéticamente predispuestos. La presencia de niveles elevados de determinadas citoquinas como TNF alfa, IL 17, 12, 23, 36 y 6, muestra a las claras el rol predisponente en el desarrollo y mantenimiento del proceso y el potencial terapéutico de éstas.
El artículo, el primero de una serie mayor de educación médica continua pone el foco en el papel central de las citocinas inflamatorias y otros factores contribuyentes, como la genética, las hormonas y los microorganismos patógenos. Las especies predominantes son el Corynebacterium (aerobios grampositivos con capacidad oportunista de infiltrarse en piel dañada); el Porphyromonas (anaerobios gramnegativos con posibilidad de prosperar en comunidades polimicrobianas en ambientes inflamatorios) y diferentes tipos de Peptoniphilus (anaerobios grampositivos que se ven comúnmente en la piel de los diabéticos o en infecciones que involucran tejidos blandos, huesos, articulaciones o cirugía). Porphyromonas y Peptoniphilus son especies que se han asociado con heridas crónicas y, por lo tanto, pueden afectar la cronicidad de HS.
Hallazgos Clínicos.
Se presenta con un curso clínico variable. Una de las principales manifestaciones de la enfermedad es la ocurrencia en áreas intertriginosas, sin embargo, puede afectar otras áreas. En orden de importancia éstas son: inguinal, axilar, perianal y, además, sub- e intermamaria en mujeres, nalgas, monte pubiano, c.c. y pabellones auriculares y párpados.
El estadio inicial no se diagnostica, sino como foliculitis. Los nódulos transitorios o persistentes, abscesos y, finalmente, cicatrices, forman parte de la definición y, estos, pueden gradualmente formar placas. 20-30% de los pacientes, también, pueden estar afectados por enfermedad o sinus pilonidal.
Diagnóstico.
Para esta enfermedad crónica e invalidante, el screening eficiente en la población, en la atención primaria programada para detectar HS en estadios iniciales, es esencial. Esto se refleja en el tiempo medio prolongado de diagnóstico, que actualmente es de 7 años. Como no existe ningún test diagnóstico específico en la atención primaria ni en dermatología, implementar los criterios diagnósticos es de alta significancia.
Evidencias recientes sugieren que una respuesta positiva a las siguientes preguntas puede identificar pacientes con HS con una sensibilidad del 90% y una especificidad del 97%: Ha tenido Ud brotes de forúnculos en los últimos 6 meses con un mínimo de 2 lesiones en una de las siguientes 5 localizaciones: axilas, ingles, genital, submamario u otras (perianal, cuello, abdomen)?. El término “forúnculo” es utilizado cuando los pacientes refieren que sus lesiones son esas. Dicha definición es muy fácil de utilizar en la atención primaria programada y ha mostrado adecuada sensibilidad y especificidad, lo que es recomendado para ser utilizado en la identificación temprana de pacientes.
Atención Primaria.
Brotes de forúnculos durante los últimos 6 meses, con un mínimo de 2 lesiones en alguna de las siguientes localizaciones: axilas, ingles, genital, perianal, cuello, abdomen, submamario.
Obligatorios.
Historia: Lesiones recurrentes dolorosas o purulentas, en más de 2 ocasiones en los últimos 6 meses.
Localización: Axila, ingle, perineo, glúteos, sub e intermamaria.
Signos clínicos: Lesiones primarias tipo foliculitis, abscesos.
Lesiones secundarias: tipo quísticas, fístula/sinus (exudativas o no exudativas), pseudocomedones dobles, cicatrices (atróficas, en banda, eritematosas, hipertróficas, lineares).
Adicionales.
Historia familiar positiva para HS.
Microbiología. Sin evidencia de patógeno o presencia de microflora cutánea normalmente en el tipo de lesión predominante.
Diagnóstico de HS.
Presencia de 3 criterios obligatorio.
Una o más localizaciones obligatorias están involucradas.
Uno o más tipos de lesiones obligatorias están presentes (nódulos, abscesos, fístula/sinus, cicatriz).
El diagnóstico de HS requiere examinación visual simple de todas las áreas del cuerpo en las cuales la enfermedad puede afectar, para esto los pacientes deben ser desvestidos.
PGA- HS.
Clear (Score 0). Ausencia de abscesos, fístulas con drenaje, nódulos inflamatorios, nódulos no inflamatorios.
Mínimo (Score 1). Ausencia de abscesos, fístulas con drenaje, nódulos inflamatorio, con presencia de nódulos no inflamatorios.
Leve (Score 2). Ausencia de abscesos y fístulas. Presencia de 1-4 nódulos inflamatorios, o 1 absceso o fístula con drenaje y nódulos no inflamatorios.
Moderado (Score 3). Ausencia de abscesos y fístulas. Presencia de más de 5 nódulos inflamatorios, o 1 absceso o fístula con drenaje y nódulos no inflamatorios.
Severo (Score 4). Presencia de 2-5 abscesos o fístulas con drenaje y más de 10 nódulos inflamatorios.
Muy Severo (Score 5). Más de 5 abscesos o fístulas con drenaje.
Diagnóstico Diferencial.
Infección estafilocóccica (lesiones purulentas diseminadas)
Enfermedad de Crohn cutánea.
Abscesos (usualmente única).
Tumores, primarios o metastásicos.
Linfogranuloma venereo.
Enfermedades raras: actinomicosis, TBC cutis colicuativa.
Marcadores de laboratorio.
No existen marcadores específicos. Los pacientes con HS activa pueden tener incremento de células, ERS acelerada y PCR +. En presencia de signos adicionales de inflamación, incluyendo fiebre debe excluirse infección de tejidos blandos.
Diagnóstico por imágenes.
La Ecografía de Alta Resolución es apropiada para evaluar los límites del absceso y la formación de fístulas, en la parte del folículo.
RMN es una alternativa para la detección de fístulas, especialmente aquellas que también involucran otros órganos (fístula dermointestinal y compromiso genito- anal). La extensión de la inflamación puede ser evaluada por termografía de la piel.
Clasificación y Evaluación de la Severidad.
La Estadificación de Hurley y PGA de HS son medidas que pueden ser utilizadas para clasificar y evaluar la severidad de la enfermedad.
Hurley divide la enfermedad en 3 estadios y es propuesta como herramienta para facilitar la decisión de tratamiento racional para un posible enfoque quirúrgico en determinadas localizaciones corporales. No es útil para enfocar tratamiento médico.
El PGA HS incluye 6 estadios con guías claras para evaluar severidad de la enfermedad y es adecuado para un seguimiento dinámico.
ESTADIFICACION DE HURLEY.
Lesiones individuales primarias y/o quistes sin fístulas ni cicatrices.
Lesiones individuales primarias y/o quistes con presencia de fístulas y cicatrices.
Lesiones primarias y secundarias confluentes en el compromiso de la superficie con fístulas y cicatrices.
Actualmente existe una actualización a esta, que clasifica aún a las etapas I y II en leve, moderada y severa.
PGA- HS.
Clear (Score 0). Ausencia de abscesos, fístulas con drenaje, nódulos inflamatorios, nódulos no inflamatorios.
Mínimo (Score 1). Ausencia de abscesos, fístulas con drenaje, nódulos inflamatorio, con presencia de nódulos no inflamatorios.
Leve (Score 2). Ausencia de abscesos y fístulas. Presencia de 1-4 nódulos inflamatorios, o 1 absceso o fístula con drenaje y nódulos no inflamatorios.
Moderado (Score 3). Ausencia de abscesos y fístulas. Presencia de más de 5 nódulos inflamatorios, o 1 absceso o fístula con drenaje y nódulos no inflamatorios.
Severo (Score 4). Presencia de 2-5 abscesos o fístulas con drenaje y más de 10 nódulos inflamatorios.
Muy Severo (Score 5). Más de 5 abscesos o fístulas con drenaje.
Evaluación de la Efectividad del Tratamiento Antiinflamatorio.
La evaluación de la respuesta clínica fue recientemente desarrollada y validada para determinar la efectividad del tratamiento antiinflamatorio. Requiere el conteo de las lesiones nodulares inflamatorias, abscesos y fístulas con drenaje al inicio y luego del tratamiento.
El éxito del tratamiento se considera con (1) al menos 50% de reducción en el número de abscesos y nódulos inflamatorios, (2) no aumento en el número de abscesos y (3) no incremento en el número de fístulas con drenaje desde inicio. Esto se correlacionó significativamente con mejoría en todos los resultados, tanto para los médicos como para los pacientes.
Este Score fácil de utilizar y se recomienda para la práctica clínica diaria, en relación con la evaluación objetiva y uniforme de los efectos del tratamiento antiinflamatorio y mejorar la evidencia basada en la práctica clínica.
Complicaciones – Comorbilidades.
En HS severa o crónica, complicaciones tales como erisipela, infección de tejidos blandos seguido de sepsis, linfangitis (elefantiasis, especialmente en área genital) y, aunque raramente, malignidad (CEC, en él área genital en hombres, con riesgo de 1.7-3.2% y un rango de mortalidad superior al 50%). La cicatrización puede generar restricción en el movimiento de las articulaciones (especialmente en axilas). Cuando la afección es anogenital, pueden observarse estricturas en la uretra, el ano y recto y fístulas parauretrales y pararectales. Puede ocurrir anemia, hipoproteinemia y/o amiloidosis cutánea.
HS se asocia a otras enfermedades como pioderma o artritis. Reduce significativamente la calidad de vida, depresión, estigmatización, inactividad, invalidez laboral y trastornos de la salud y la actividad sexual.
Estos pacientes presentan un fuerte trastorno psicológico frecuentemente, como resultado en la restricción de los contactos sociales, incluso con desarrollo de depresión. El 75% de los pacientes afectados son fumadores y, de ellos, gran proporción son obesos.
El desarrollo de factores de riesgo CV es frecuente, tales como tabaco, obesidad, dislipidemias, DBT y síndrome metabólico, significativamente mayor que los controles sanos. Otros procesos relacionados son las tiroideopatías y la enfermedad de Crohn.
Enfermedades dermatológicas se encuentran entre las comorbilidades, como acné conglobata, celulitis disecante de cuero cabelludo, quiste o sinus pilonidal y acné queloideano de la nuca, con quienes constituye la tríada o tétrada de oclusión folicular.
Por lo tanto, la evaluación de inicio de estas enfermedades asociadas, con escalas apropiadas como DLQI y escala visual analógicas, sólo con evaluación de la presencia de factores de riesgo CV pueden facilitar la detección temprana y la implementación a tiempo, de estrategias para modificarlos o prevenirlos.
Tratamiento.
Bibliografía. Goldburg, S.R.; Strober, B.E.; Payette, M.J. Hidradenitis suppurativa: Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2020; 82(5):1045-1058.
El vitiligo es un desorden pigmentario adquirido, autoinmune, benigno, de curso indolente, constituida por la aparición de lesiones maculosas acrómicas (blancas) en cualquier sector del tegumento, con alto impacto psicosocial en el paciente, frecuentemente asociado a otros procesos autoinmunes como problemas tiroideos, diabetes o enfermedad celíaca.
DIAGNÓSTICO.
Es un proceso de diagnóstico eminentemente clínico, aunque en algunas ocasiones puede ser necesaria la biopsia para la diferenciación con otros procesos pigmentarios.
ALGUNAS IDEAS QUE PUEDEN AYUDAR AL PACIENTE.
No esconda su problema. No deje de visibilizar su problema, exteriorícelo, puede ayudarlo psicológicamente, a aceptar su enfermedad y a convivir de una manera más amigable con ella. Sepa que 2 a 3 de cada 100 personas la padecen igual que usted, no se encuentra aislado.
Busque ayuda. Existen muchas alternativas de tratamiento, los médicos especialistas conocemos su enfermedad e investigamos distintas maneras de ayudarlo.
No se automedique. No utilice alternativas que no se han demostraron efectivas, consulte con un médico dermatólogo, porque el vitiligo pertenece a su territorio específico y lo conoce. No permita que personas sin conocimientos específicos y sin títulos profesionales le indiquen soluciones mágicas.
Es sólo una mancha. El vitiligo no afecta órganos internos, no es cáncer, no genera riesgo de vida, pero si puede avisar sobre la posibilidad de estar padeciendo otras enfermedades, por lo que es necesario acudir al médico especialista, quien no sólo dará tratamiento a su enfermedad, además investigará sobre posibles comorbilidades.
CLINICA.
Se presenta como manchas blancas o despigmentadas, bien delimitadas, redondas, ovales o lineales, cuyo tamaño puede rondar desde milímetros a centímetros, con el tiempo pueden crecer centrifugamente de modo impredecible. Puede afectar más frecuentemente manos, antebrazos, pie y cara y, en esta, perioral y periorbicular.
Las variantes clínicas son:
Vitiligo Tricrómico. Caracterizado por un halo hipopigmentado, entre un área acrómica central y la periferia pigmentada. El área intermedia evoluciona hacia despigmentación total. Esto resulta en 3 tonos de piel. Su ocurrencia depende del tipo de piel del paciente.
Vitiligo Inflamatorio Marginal. Una variedad muy infrecuente, que presenta un área externa eritematosa pruriginosa.
Vitiligo Cuadricrómico. Una variante que refleja la aparición de un área de color marrón oscuro en región perifolicular en situación de repigmentación.
Esta enfermedad también puede clasificarse en dos grandes grupos: segmentarías y no segmentario. Aunque también es posible clasificarlo en localizado, cuando afecta una sóla área del cuerpo, o diseminando, cuando compromete más de un área corporal. Pero la primera de ellas es la más utilizada por los médicos.
Vitiligo Segmentario. Esta forma presenta lesiones típicas que transcurren a través de una de las líneas de Blaschko. Es unilateral, y nunca atraviesa la línea media del cuerpo. Generalmente es de inicio temprano y progresa rápidamente en el área afectada. Aunque este puede detenerse y permanecer estable a lo largo del tiempo. Esta variedad no se asocia a trastornos tíroideos u otras enfermedades autoinmunes.
Vitiligo No Segmentario. Este un término que engloba a todas las variantes de Vitiligo que no sean segmentarias. Estas variantes están más asociadas a autoinmunidad e inflamación, tal es el caso de formas como el halo nevus.
TRATAMIENTO ORAL.
CORTICOSTEROIDES.
En varios estudios con gran número de casos, con régimen oral minipulso (OMP) con Betametasona/Dexametasona 2.5-5mg/día, dos días seguidos, por semana, con una duración de 2-6 meses, en cuadros de Vitiligo inestable, se logró interrumpir su progresión en más del 80%, y se consiguió signos de repigmentación en la misma proporción.
CICLOSPORINA.
Las series de casos que probaron Ciclosporina, lo hicieron con esquema clásico de 3mg/kg, por 12 semanas, observándose una progresión del 61% y mejoría evidente del Vitiligo Área and Severity Index (VASI).
METOTREXATE.
Hay muy poca evidencia al respecto, pero en un estudio abierto de 50 casos, la dosis utilizada es de 10mg/semana, asociado a minipulso oral de corticosteroides (OMP) con dexametasona 2.5mg/bisemanal. Puede utilizarse asociado con OMP o sólo cuando los corticosteroides están contraindicados. Se ha visto una reducción del Vitiligo Disease Activity (VDA).
MINOCICLINA.
Es un antibiótico de la familia de las tetraciclinas, tiene propiedades antiinflamatorias e inmunomoduladoras y, además, posible rol en la destrucción de radicales libres, lo que podría sugerir su capacidad de salvar al melanocítico de su destrucción por el estrés occidativo in vitro.
Se ha observado el freno en el avance de la enfermedad en un grupo de 32 pacientes con 100mg/día. Con este esquema se ha observado reducción en VDA y en VASI con significativa repigmentación.
Sin embargo, en estudios prospectivos, randomizados comparativos se observó mayor eficacia repigmentante con UVB-NB, y obtención de la estabilidad de la enfermedad en el tratamiento de Vitiligo inestable (76.1% con UVBnb vs 33.3% con minociclina). Esto demuestra que la minociclina es inferior en efectividad a UVB, pero es importante saber que la minociclina monoterapia es comparable a corticosteroides OMP, y puede ser de gran ayuda cuando la fototerapia UVB no es factible.
AFAMELANOTIDE.
Es un análogo de la hormona alfa-melanocito estimulante, uniendo al receptor de la melanocortina-1, estimulando la melanogenesis, siendo usada con adyuvante de fototerapia. Esta asociación es un promotor de la estimulación y la proliferación melanocítica. En el estudio más importante realizado hasta el momento, donde se asoció UVB-NB con Afamelanotide en implante subcutáneo de 16mg, la repigmentación fue evidentemente superior a aquella como monoterapia, siendo esto más visible en fototipos IV-VI.
Los efectos adversos más frecuentes son eritema, prurito, náuseas, cefaleas e hipercromia de la piel no afectada.
INHIBIDORES DE JANUS KINASA.
La escasa información existente los muestra como adyuvantes para mejorar la respuesta repigmentante de la fototerapia. INF-gamma induce la expresión de CXCL-10 en queratinocitos, ha sido propuesto como intermediario en la despigmentación en vitiligo. Todo esto se produce a través de JAK. Por todo esto, se ha postulado a Tofacitinib para inhibir la expresión de INF-gamma, y reducir de esta manera, la expresión de CXCL-10, resultando en repigmentanción.
Los escasos resultados positivos, la inaccesibilidad por sus costos y la posible carcinogenesis asociada los relega ante otras alternativas de tratamiento.
Generalidades
La etiología de estas enfermedades aún hoy permanece desconocida, habiendo conceptos bien definidos. La predisposición genética es indispensable para su desarrollo, siendo los gatillos mecanismos de autoinmunidad.
Se relaciona al Penfigoide con ciertos fármacos con furosemida, antiinflamatorios no esteroides y algunos antibióticos. Pero desde hace años que se sostiene su relación con ciertos procesos neurológicos como esclerosis múltiple, demencia o el párkinson, especialmente en la variante localizada en extremidades inferiores, más frecuente en mujeres añosas.
FISIOPATOGENIA
En el Penfigoide se producen de modo aberrante dos autoanticuerpos BP180 y BP230, dirigidos contra antígenos de membrana, responsables de la cohesión dermoepidérmica, la cual se pierde por la infiltración eosinofílica.
En el Pénfigo, los anticuerpos están dirigidos contra proteínas Desmogleina 1 y 3, constituyentes de los desmosomas, responsables de la adhesión intercelular. En ambos casos, la pérdida de la coherencia resulta en la formación ampollar.
Inmunofluorescencia Directa.
Esta es, tal vez, la técnica que permite el diagnóstico de certeza, considerada como técnica estándar de oro. Consiste en obtener una muestra de piel sana, de una zona cercana a una lesión mediante técnica de punch o losange. Esta muestra se cultiva en solución con reactivo inmunomarcado con reactivo fluorescente. En el caso de Penfigoide Ampollar se obtiene una marcación a nivel de unión dermoepidérmica, de una imagen granular a expensas de IgG/C3, patrón que puede ser compartido con la Epidermolisis Ampollar, para lo cual debe asociar la técnica de spleet para su diferenciación. En el caso de Pénfigo Vulgar la marcación se produce a nivel intraepidérmico, supra basal (imagen en panal de abejas), también a expensas de IgG/C3. Hay una variante de Pénfigo conocida como IgA, por su marcación.
PRONÓSTICO.
En el caso del Penfigoide Ampollar en curso tiende a ser autolimitado, durando de meses a años, en tanto sea causado por algún factor identificable, como los fármacos antes mencionados. En el caso del Pénfigo Vulgar, el curso puede ser más prolongado y no ser autolimitado, salvo en caso de la variante paraneoplásica, donde resuelve en caso de resolver el proceso subyacente. Ha sido conocido su desenlace fatal irremediablemente hasta la aparición de los corticosteroides, mortalidad que ha descendido dramáticamente luego de su aparición, sin dejar de considerar su alta morbilidad.
DIAGNOSTICO Y CONCEPTOS GENERALES.
Desorden cutáneo, crónico, polimorfo, muy pruriginoso, inducido por trastornos en la absorción del gluten, que presenta como principales carácterísticas depósitos de IgA granular subepidérmico y grados variables de enteropatía por gluten, idéntico a lo observado en la Enfermedad Celíaca (EC), por lo que puede combinar características de la enfermedad cutánea y la intestinal.
Su primera descripción corresponde a Louis Duhring en el año 1884, como un proceso crónico de piel, intensamente pruriginoso y polimorfismo característico, y su asociación con trastornos intestinales con alteraciones de la absorción del gluten, recién ocurre en el año 1966 y lo realiza Marks y colaboradores. Cormane, en 1967, describe la presencia de una inmunoglobulina en la unión dermoepidérmica que, en 1969, van der Meer identifica como IgA. Chorzelski, en 1979, diferencia la Dermatitis Herpetiforme (DH) de la Enfermedad IgA lineal en base a los hallazgos en la estudios de inmunofluorescencia y en el año 1983 también describen el hallazgo de un autoanticuerpo contra endomisio presente tanto en suero de pacientes con DH como de EC.
BASES GENÉTICAS.
Se encuentra estrictamente asociado a HLA-DQ2 y DQ8. En la población caucásica, el 85% se asocia a DQ2 y el 15% restante a DQ8.
EPIDEMIOLOGÍA.
DH es una enfermedad rara, con una prevalencia de 10-75/100000 hab, y una incidencia de 1-3.5. De muy baja ocurrencia en la población asiática y afro-americanos, más frecuentes entre los noreuropeos y Estados Unidos. Mientras el inicio de la enfermedad en la adultos es más frecuente en Europa del norte, la afección de los niños en más frecuente en los países mediterráneos. La mayor prevalencia de 75/100000 y la mayor incidencia de 3.5% se registra en Finlandia. Respecto de los géneros, mientras EC es más frecuente en mujeres, se observa mayor frecuencia de DH en varones, siendo el rango V:M de 3:2. Pero es cierto que las mujeres se ven más afectadas por debajo de los 20 años.
DIAGNÓSTICO Y CONCEPTOS GENERALES.
Es un desorden sistémico inmunomediado, relacionado con la mala absorción de determinados alimentos que contienen gluten, en personas genéticamente determinadas, caracterizado por la combinación variable de manifestaciones clínicas dependientes del gluten, anticuerpos celiacos específicos, HLA DQ2 y DQ8 y enteropatía. El gluten ingerido por trigo, avena, cebada y centeno dispara la activación de células T, produciendo auto anticuerpos contra transglutaminasa, atrofia de vellosidades intestinales con hiperplasia de las criptas.
CARACTERÍSTICAS COMUNES A EC Y A DH.
Es un desorden sistémico inmunomediado, relacionado con la mala absorción de determinados alimentos que contienen gluten, en personas genéticamente determinadas, caracterizado por la combinación variable de manifestaciones clínicas dependientes del gluten, anticuerpos celiacos específicos, HLA DQ2 y DQ8 y enteropatía. El gluten ingerido por trigo, avena, cebada y centeno dispara la activación de células T, produciendo auto anticuerpos contra transglutaminasa, atrofia de vellosidades intestinales con hiperplasia de las criptas.
Los mismos péptidos derivados del gluten (principalmente gliadina) pueden ser el gatillo en ambas entidades y la presentación a la célula T requiere la presencia de HLA DQ2 y 8. Los autoanticuerpos hacen blanco en los mismos epitopes de Antitransglutaminasa 2 (TG2) produciéndolos durante el consumo de gluten, mientras que dejan de producir cuando se suspende el consumo.
En pacientes con DH puede verse el cuadro completo de enteropatía por gluten: 25-30% de los casos presentan arquitectura vellositaria conservada (MARSH 0) o con aumento de linfocitos intraepiteliales (MARSH I), hiperplasia de las criptas (MARSH II), mientras que en 70-75% de los pacientes, además se observa atrofia vellositaria moderada a severa con hiperplasia de criptas (MARSH IIIC). Pacientes con DH conocida que presentan estructura vellositaria normal, se altera luego de consumir gluten. Tanto las manifestaciones gastrointestinales como cutáneas en estos pacientes remiten con dieta estricta libre de gluten. La intolerancia al gluten es definitiva tanto en DH como en EC y no tiene curación. Manifestaciones de ambas enfermedades pueden alternar en una misma persona en diferentes momentos a lo largo de su vida y en familiares de primer grado.
El diagnóstico de DH se basa clásicamente en muy pocos elementos que se detallan a continuación.
Historia Clínica.
Examen Físico.
Examen Histopatológico.
Inmunofluorescencia Directa.
Examen Serológico (Inmunofluorescencia Indirecta).
Evaluación Gastrointestinal.
Es importante considerar no indicar tratamiento específico con dietas libre de gluten y/o dapsona hasta completar los procedimientos diagnósticos específicos ya pueden modificar los resultados y dificultar las conclusiones.
Debido a la predisposición genética de la DH, es importante realizar screening en todos los familiares genéticamente relacionados buscando DH o EC.
Manifestaciones Cutáneas. Es característico de esta enfermedad su polimorfismo de sus manifestaciones cutáneas, mostrando papulas, excoriaciones, vesículas y ampollas pequeñas, ronchas, todas ellas en distintos estadios de evolución, asociadas a prurito severisimo e incoercible y refractario a cualquier medida terapéutica que, es cercano a una sensación urente, ubicada preferiblemente en áreas extensoras del cuerpo, frecuentemente expuestas a roce, como codos, rodillas, hombros, espalda y cuero cabelludo. Es muy importante su impacto en la calidad de vida provocando alteraciones psicológicas a gran escala. Como consecuencia del rascado crónico, se observan lesiones muy comunes como liquenificación, hiper e hipoccomía residual. El cuadro puede iniciar con petequias y púrpura de distribución acral. Lesiones hemorrágicas en dedos de manos y pies.
Formas raras de presentación pueden ser compromiso facial exclusiva, con lesiones maculares, apariencia simil vasculitis leucocitoclástica, queratodermia palmoplantar, lesiones urticarianas o tipo prurigo..
Manifestaciones Orales: El compromiso oral es sumamente infrecuente, pero puede acompañarse de síntomas subjetivos como boca seca o sensación urente. En el examen físico puede observarse aftas, úlceras, máculas rojas en mucosa oral y lengua. .
Aquellos pacientes con manifestaciones gastrointestinales presentan más compromiso mucoso que aquellos con manifestaciones cutáneas solamente.
Manifestaciones Gastrointestinales y Otras.
Forman parte del espectro de manifestaciones de DH, aunque su presencia es menos frecuente que en EC. Estas pueden ser asintomáticas a pesar del hallazgos de cambios histológicos. Muy pocos pacientes (15-20%) pueden presentar diarrea crónica o episódica, constipación, retortijones, dolor, pérdida de peso o palidez. En niños puede registrarse mala absorción, déficit de hierro y reducción de los índices de crecimiento.
Otras manifestaciones Aisladas.
Otros hallazgos posibles en estos pacientes incluyen infertilidad, enfermedad hepática, neuropatía, neuropatía y ataxia cerebelosa.
Además deben evaluarse posibles enfermedades asociadas como tiroideopatias, malignidad (linfomas, leucemias), diabetes, enfermedad de Adisson, vitiligo, alopecia areata y otras enfermedades autoinmunes.
Histopatología.
La muestra para biopsia debe ser obtenida con un punch de entre 4 y 5mm, de una lesión ampollar intacta y evitar que ésta se rompa para su correcto estudio. Los elementos diagnósticos son acúmulos de neutrófilos conformando microabscesos en dermis papilar. Despegamiento dermoepidérmico con la consecuente formación de vesícula. Presencia de eosinófilos en número varíable en el infiltrado con predominio de los neutrófilos. Estos hallazgos por sí sólos no permiten diferenciarla de otras enfermedades inmunoampollares como enfermedad IgA lineal, penfigoide ampollar o epidermolisis ampollar adquirida, entre otras.
Inmunofluorescencia Directa..
Esta es, tal vez, la técnica que permite el diagnóstico de certeza, considerada como técnica estándar de oro. Consiste en obtener una muestra de piel sana, de una zona cercana a una lesión mediante técnica de punch o losange. Esta muestra se cultiva en solución con reactivo inmunomarcado con reactivo fluorescente. En el caso de DH se obtiene una marcación a nivel de unión dermoepidérmica, de una imagen granular a expensas de IgA, patrón patognomómico. Además los depósitos de IgA se observan en los vasos de dermis papilar, a veces también reticular y, menos, frecuente, fibras elásticas, músculo piloerector, alrededor de folículo piloso y en membrana basal de ductus y glándula sudorípara.
En ocasiones se han visto series de pocos pacientes que presentan depósitos granulares de C3 únicamente a nivel dermoepidérmico, en ausencia de otros marcadores, proponiéndose la denominación de “Dermatosis C3 granular”.
Se observan rara vez falsos positivos en el caso de EC en ausencia de hallazgos de DH, tal es el caso de dermatitis por contacto, tinea o psoriasis.
También son infrecuentes de ver los falsos negativos, por razones de errores de técnicas, mala pepraración de la muestra y defecto de la toma de muestra. También puede observarse en pacientes medicados con corticosteroides durante la toma.
Inmunofluorescencia Indirecta (IFI).
Los anticuerpos investigados son antigliadina, antigliadina deaminada, antiendomisio y antitransglutaminasa. La misma puede realizarse por IFI o por ELISA, sólo importándonos la primera de ellas.
La microscopía por IFI sirve para detectar cuali-cuantitativamente anticuerpos antiendomisio (EMA) en el suero de estos pacientes, teniendo una doble ventaja: diagnóstico y screening, pero es un método sumamente complejo y requiere personal capacitado. Para detección de Anticuerpo Antiendomisio IgA (IgA EMA) se utiliza esófago de mono con un patrón en “pañal de abeja” alrededor de las fibras de músculo liso. Otros sustratos pueden ser utilizados como utero de mono, esófago de conejo, apéndice humano normal, etc. En pacientes con DH no tratados, EMA presenta una sensibilidad cercana a 60-90%, mientras que en pacientes con EC es aún mayor.
Diagnóstico Intestinal.
Se recomienda la biopsia intestinal en los pacientes con DH para evaluar el grado de enteropatía presente. Deben realizarse cuatro tomas biopsia de duodeno y una de bulbo duodenal para la investigación por vídeo Endoscopia alta.
Los test de malaabsorción intestinal tiene muy baja sensibilidad, ya tiene que haber una muy severa disminución de la superficie vellositaria para mostrar la alteración de la absorción. Mientras tanto, bajos niveles de ferritina sérica puede ser signo de mala absorción. Si bien el diagnóstico de DH se realiza por IFD de piel solamente, la evaluación de intestino delgado es necesaria para generar una estrategia de tratamiento, dejando en claro que que no es solamente un problema de piel.
Exámenes de Laboratorio Auxiliares.
Genotipificación de Haplotipos HLA.
La asociación estrecha con EC, requiere que se realice la tipificación de haplotipos HLA como mayor factor de riesgo genético, siendo el más frecuente el heterodimero DQ2,5, DQ2,2 también conocido como DQ2 (95%) y, en menor medida, el DQ8 (5%), que proveen una sensibilidad cercana al 100% para EC y DH.
Investigación de Comorbilidades.
La asociación estrecha con EC, requiere que se realice la tipificación de haplotipos HLA como mayor factor de riesgo genético, siendo el más frecuente el heterodimero DQ2,5, DQ2,2 también conocido como DQ2 (95%) y, en menor medida, el DQ8 (5%), que proveen una sensibilidad cercana al 100% para EC y DH.
Múltiples enfermedades autoinmunes como tiroideopatias, diabetes tipo I, enfermedad de Addison, síndromes multiendocrinos y trastornos linfoproliferativos se observan con mayor prevalencia en DH y EC.
La enfermedad tiroidea subclínica es muy frecuente en estos pacientes y debe ser testeada siempre. En el caso de los linfomas es menos frecuente la asociación por ende, la búsqueda debe realizarse sólo ante la sospecha clínica.
La evaluación del riesgo cardiovascular debe ser considerada ya que la alimentación de estos pacientes reemplaza los déficit nutrionales con exceso de lípidos, azúcar y sal, transformando en una dieta hipercalórica, con el consecuente impacto cardiometabólico (obesidad, dislipemia, insulina resistencia, síndrome metabólico y ateroesclerosis, por lo que en hombres mayores de 50 años y mujeres de 50 años, debe hacerse foco en estos factores de riesgo, como tabaco, alcohol, hipertensión, diabetes, dislipemia e hiperuricemia.
Evaluación de Malaabsorción.
Las principales consecuencias de no poder incorporar nutrientes correctamente son pérdida de peso, alopecia, anemia y deficit de absorción de vitaminas y minerales. Es necesario realizar una evaluación correcta de los aportes nutricionales al inicio de la evaluación de la DH. La mejor evaluación de la absorción intestinal es el estudio histopatológico de las vellosidades intestinales. Otros test utilizados para esta evaluación (D-Xilosa) no son tan sensibles en la detección de enteropatía. Estudios que evalúen la densidad mineral ósea, pueden alertar sobre mala absorción.
Diagnóstico Diferencial.
Las principales consecuencias de no poder incorporar nutrientes correctamente son pérdida de peso, alopecia, anemia y deficit de absorción de vitaminas y minerales. Es necesario realizar una evaluación correcta de los aportes nutricionales al inicio de la evaluación de la DH. La mejor evaluación de la absorción intestinal es el estudio histopatológico de las vellosidades intestinales. Otros test utilizados para esta evaluación (D-Xilosa) no son tan sensibles en la detección de enteropatía. Estudios que evalúen la densidad mineral ósea, pueden alertar sobre mala absorción.
DH debe diferenciarse de todas aquellas dermatosis pruriginosas, escoriadas y ampollares. Pero las más importantes son las inmunoampollares. Aquí fundamentalmente encontramos penfigoide ampollar, epidermolisis adquirida, dermatosis IgA lineal, pénfigo IgA, lupus eritematoso ampollar, penfigoide anti Laminina (o anti p-200), pénfigo herpetiforme.
De todos modos, debido a su gran polimorfismo, puede confundirse con otras dermatosis pruriginosas, escoriadas, no autoinmunes, como dermatitis atópica, escabiosis, foliculitis, prurigos, reacción por picaduras de artrópodos.
La búsqueda debe ser permanente y realizar todos los estudios complementarios necesarios, ya que presentación clínica puede ser muy conflictiva.
Tratamiento.
La principal opción terapéutica en la actualidad es la implementación de la dieta libre de gluten (DLG) asociado a Dapsona. La DLG es la única opción causal, que sólo puede iniciar luego de completar los exámenes.
Otras opciones terapéuticas son sulfazalasina, corticosteroides a altas dosis y antihistamínicos, aunque menos eficientes, se consideran ante la imposibilidad de ser administrada la dapsona.
DLG se asocia con una rápida resolución de los signos y síntomas de la enfermedad celíaca, eliminación de los autoanticuerpos IgA circulantes y la disminución del riesgo de desarrollar linfoma intestinal promovido por la estimulación antigénica. Sin embargo, al resolución de las lesiones cutáneas puede necesitar meses e incluso, años. Los autoanticuerpos IgA pueden estar depositados en la dermis hasta una década, siempre bajo estricto cumplimiento de DLG. En relación a los beneficios de DLG, promueve la mineralización ósea, mejora la calidad de vida y previene la DH refractaria.
El gluten provee estructura y elasticidad a los productos de panadería. DLG consiste en eliminar cereales como trigo, avena, cebada y centeno, en productos como panificados, pastas, horneados. Los cereales prohibidos pueden ser reemplazados por otros carbohidratos complejos como arroz o maíz, y algunos pseudo cereales como sorgo, mijo, quinoa. Otros alimentos que pueden ingerirse sin restricción son huevo, lácteos, quesos, carnes rojas, pescados, frutas, vegetales y legumbres.
Cabe destacar que los pacientes no deben comenzar con una DLG antes de recibir el diagnóstico final.
Tanto las regulaciones locales como internacionales ayudan a estos pacientes a identificar los alimentos libres de gluten.
Para considerar la condición “libre de gluten” los alimentos deben contener menos de 20mg/kg de gluten. Los alimentos considerados con muy bajo contenido en gluten tienen 100mg/kg de gluten, y no son aptos para pacientes con EC/DH y su consumo debe ser restringido.
Pequeñas cantidades de gluten pueden actuar como gatillo en pacientes con DH. El gluten puede estar escondido en algunos alimentos como salsas, comidas preparadas e incluso, en pescados. Por lo tanto, la contaminación cruzada puede estar presente en comidas preparadas en casa o en un restaurante.
La eliminación de la dieta de cereales que contienen gluten puede factor causal de deficiencia de vitamina B. Ante la implementación de DLG debe indicarse el agregado de hierro y calcio.
Para formular una correcta DLG es indispensable contar con un nutricionista, que pueda proveer un listado de alimentos que contengan gluten primariamente o posiblemente contaminados. Los pacientes que siguen una dieta que se basa únicamente en el arroz y el maíz como azúcares complejos, en teoría, tienen un mayor riesgo de exposición al arsénico y a las micotoxinas.
Es muy importante que a pacientes con comorbilidades como diabetes tipo I se le informe las alternativas dietéticas como productos libres de gluten y cereales con alto índice glucémico.
Sobrepeso y obesidad afectan tanto a pacientes con EC como con DH, y se presentan al inicio de la enfermedad. Tener en cuenta que con el inicio de la DLG puede producirse un rebote en el peso del paciente, por lo que debe implementarse una dieta normocalórica con el objeto de prevenir trastornos cardiovasculares.
Durante la DLG debe considerarse el aporte de micronutrientes y suplementos vitamínicos (vitamina D, hierro, calcio, etc), que debe evaluarse según el déficit que presente. De todos modos esto en contradictorio debido a la presencia de mala absorción que afecta su incorporación.
Dapsona.
Es la droga de elección de primera línea para el tratamiento sintomático de DH. Mientras la DLG comienza a ser efectiva (6-24 meses), esta mitiga los daños causados por la enfermedad y el prurito. Su inicio de acción es rápido, resolviendo lesiones y síntomas en 3-4 días, en tanto que las manifestaciones esparcen rápidamente ante su suspensión.
Se considera de vital importancia el dosaje de glucosa 6-fosfato deshidrogenasa previo a su administración. La dosis de inicio depende de la severidad de las manifestaciones. Iniciar regímenes de bajas dosis puede minimizar el riesgo de eventos adversos. Puede iniciarse con 25mg/día por medio, para luego subir 25mg/día, cada semana hasta llegar a 200mg/día, hasta lograr el control de la enfermedad. Luego, como fase de mantenimiento, establecer dosis de 0.5-1mg/kg/día. Si el compromiso cutáneo es severo y no presenta deficiencia enzimática, puede iniciarse con 50mg dos o tres veces por día. En caso de daño renal no es necesario realizar ajuste de dosis.
El mecanismo de acción de DDS aún no está totalmente dilucidado. Presenta acción antiinflamatoria por inhibición de neutrófilos y activación y migración de eosinófilos. Previene la destrucción tisular por inhibición de mieloperoxidasa, enzima neutrofilica responsable de la producción de tóxicos oxidativos.
Los efectos adversos de DDS son habitualmente dosis-dependientes, aparecen luego de determinado tiempo y son bien tolerados en pacientes jóvenes y de mediana edad. Los idioasincráticos pueden aparecer raramente. Los efectos adversos mayores dosis dependientes (tóxicos) son metahemoglobinemia, anemia hemolítica, neutropenia, cefalea, mareos, debilidad, fatiga, náuseas y vómitos. Son más frecuentes en aquellos pacientes con deficiencia enzimática, en quienes padecen comorbilidades que disminuyen la oxigenación tisular y en personas añosas. En estas condiciones es aconsejable administrar dosis bajas y control estricto del paciente. Con niveles de metahemoglobinemia de entre 20-40%, pueden aparecen síntomas como los mencionados anteriormente, lo que requiere reducción de dosis. Con niveles superiores al 45% (dosis de 200mg/día) pueden observarse eventos adversos serios como acidosis, disnea, crisis epiléptica, arritmias y coma.
Otros eventos adversos a DDS incluyen la hipersensibilidad que se presenta asociado a eosinofilia y síntomas sistémicos (DRESS) dentro de 2-6 semanas de tratamiento, hipo-agranulocitosis, fotosensibilización, neuropatía periférica, entre otros. Si bien atraviesa placenta, se considera segura para madre y feto. Esta presente en leche materna y puede producir anemia en bebés.
Definición. Qué es el Lupus Eritematoso.
Lupus Eritematoso (LE) es una enfermedad infrecuente, inflamatoria, clásico ejemplo de autoinmunidad, tal vez la primera donde se describió este fenómeno..
En el caso de Lupus Eritematoso Cutáneo (LEC) las manifestaciones clínicas son variadas y heterogéneas. Su ocurrencia puede ser como proceso independiente o como una manifestación de Lupus Eritematoso Sistémico (LES), por lo que ambas entidades deben ser claramente diferenciadas.
Las manifestaciones cutáneas se diferencian en específicas y no específicas, de acuerdo a criterios histológicos, según James Guilliam. Las primeras fueron luego evaluadas según criterios clínicos, histológicos, serológicos y genéticos. La Clasificación de Düsseldorf (2004) es la última aceptada y es detallada a continuación:
Lupus Eritematoso Cutáneo Agudo (LECA)
Lupus Eritematoso Cutáneo Subagudo (LECSA)
Lupus Eritematoso Cutáneo Crónico (LECC)
Lupus Eritematoso Cutáneo Crónico Discoide (LECD)
Lupus Eritematoso Cutáneo Sabañón o Lupus Sabañón
Lupus Eritematoso Cutáneo Profundo/Paniculitis Lúpica
Lupus Eritematoso Cutáneo Intermitente (LECI)
Lupus Eritematoso Cutáneo Tumidus (LECT)
Entre las manifestaciones inespecíficas podemos encontrar al fenómeno de Raynaud, telangiectasias periungueales, livedo racemoso, entre otros.
Lupus Eritematoso Cutáneo Crónico (LECC).
El LECD es, tal vez, la variante más característica y frecuente. Puede presentarse como localizada (cara y cuero cabelludo) o diseminada (frecuentemente asociada a LES). Puede localizarse también en tronco superior y cara extensora de extremidades y, menos frecuente, puede afectar la mucosa oral.
Constituido por placas eritematosas, hiperqueratósicas, adherentes. Su remoción causa dolor. Su margen es eritematoso e hiperpigmentado, con centro atrófico e hipopigmentado. Alopecia cicatrizal en áreas pilosas (cuero cabelludo). Lesiones discoideas en áreas de labios. Mutilaciones en nariz y boca (cicatriz perioral vermicular). Aunque infrecuente, puede desarrollar carcinoma espinocelular en áreas cicatrizales, luego de varios años de desarrollado..
En el 10% de los casos evoluciona a LES.
Lupus Eritematoso Sabañón.
Presenta con procesos inflamatorios, dolorosos a la presión, como grandes nódulos, con depresión central o ulceración.
Los sitios de predilección coincide con sabañones, en áreas acrales, dedos de manos y pies, pabellón auricular y punta nasal.
Su frecuencia aumenta en temporadas frías y húmedas.
Serología: la positividad para ANA, AntiRo y factor reumatoideo es variable. La presencia de AntiADN es inusual.
La asociación con LES es del 20%.
Lupus Eritematoso Profundo.
También conocido como paniculitis lúpica. De localización subcutáneo, es nodular o discoide, con infiltración firme, superficie eritematosa inflamatoria, y puede superponerse con lesiones discoides. Puede localizarse en glúteos, caderas, muslos, cara, tórax y antebrazos. En raras ocasiones puede debutar con edema periorbitario. La ulceración y la calcificación son eventos posibles de hallar, y su curación puede resultar en cicatrices y lipoatrofia profunda.
Serología: ANA + en más del 75%. No presenta positividad para AntiADN, si esto ocurre es evidencia de transición a LES.
El 35-50% de los casos asocia a LES.
Lupus Eritematoso Tumidus.
Las lesiones clásicas son placas eritematosas, infiltradas, urticarianas con superficie suave, sin compromiso epidérmico, con patrón anular o semicircular.
Se distribuye especialmente en áreas expuestas al sol, cara, tórax superior, extremidades superiores.
Estas lesiones resuelven con cicatrices o secuelas pigmentarias.
Presenta fotosensibilidad exagerada.
Serología: ANA (+) en 10-30%, AntiRo/La: (+) 5%.
Su curso tiene muy buen pronóstico, con remisión espontánea frecuentemente.
Etiopatogenia.
La patogenia de la LEC es compleja y multifactorial, incluida la susceptibilidad genética, los desencadenantes ambientales y la participación de los sistemas inmunitarios innato y adaptativo. La patogenia de LEC incluye el papel de la radiación ultravioleta (UV), la eliminación deficiente de células apoptóticas y complejos inmunes junto con la acumulación de autoanticuerpos y la desregulación de las células T y B.
Para poder entender determinadas terapias biológicas es indispensable saber el rol que cumple el Linfocito T en la etiopatogenia del LE. La célula T es un actor central en el LES actuando como mediador en la cascada del proceso inmunológico. Interactúa con la Célula B produciendo autoanticuerpos IgG de alta afinidad asociados con la generación del LES. Además estimula a la Célula B y a otros efectores celulares de la cascada a producir las citoquinas responsables de la actividad del LES. En el caso de LEC, la inflamación es predominantemente mediada por Th1. El IFN tipo I, producido por las células dendríticas plasmocitoides reclutan los Linfocitos Th1 hacia las lesiones epidérmicas de LEC. La correlación histológica de estos eventos epidérmicos, es conocida como dermatitis de interfase.
Epidemiología.
El viraje de LEC a LES se considera en 20% del total de los pacientes, que se produce en 3 a 5 años, desde el diagnóstico. El 30% de los pacientes con LEC desarrollan más de una variante. Habitualmente comienza entre la 3º y la 4º década de vida. La proporción por género hombre:mujer es de 3:1 en el LEC mientras es de 9:1 en el LES.
Diagnóstico.
Basado en el hallazgo de manifestaciones clínicas, el estudio histopatológico y los resultados de exámenes de laboratorio.
La evaluación de las manifestaciones cutáneas puede mejorar con el uso de los scores de medida de actividad del LES y con los específicos de LEC. Es importante determinar si las lesiones cutáneas reflejan actividad de la enfermedad, habitualmente reversible con tratamiento, en oposición al daño cutáneo irreversible (cicatrices, pérdida de cabello, hipo o hiperpigmentación). Determinar la respuesta al tratamiento en LEC puede ser dificultoso debido al gran espectro de manifestaciones cutáneas.
Contamos con varios métodos de medición de actividad en esta enfermedad, como por ejemplo: Sistemic Lupus Eritematoso Disease Activity Index (SLEDAI), British Isles Lupus Assessment Group (BILAG) index, y el SLEDAI 2K RI50, cuando estamos frente a un paciente con LES; en caso de LEC, Cutaneous Lupus Area and Severity Index (CLASI) es la herramienta utiliza para evaluar actividad, severidad, daño y respuesta al tratamiento.
Histopatología.
Ante la sospecha de LEC debe realizarse biopsia de una lesión activa libre de tratamiento alguno. Para arribar al diagnóstico, en esta muestra deben encontrarse los siguientes elementos: dermatitis de interfase con infiltrado linfocitario epidérmico, vacuolización de queratinocitos basales y cuerpos coloides. Elementos secundarios pueden estar presentes, como acantosis, infiltrados dérmicos y depósitos de mucina, de modo variable, dependiendo de la variante clínica. Para el hallazgo de estos elementos debemos contar con tinciones especiales como PAS para membrana basal y células dendríticas plasmocitoides. AIcian blue para la detección de depósitos de mucina en dermis.
Inmunofluorescencia Directa..
En LEC muestra depósitos granulares de C3, IgG e IgM en piel lesional. Puede ser diagnóstico de certeza. Pueden verse falsos-positivos áreas expuestas al sol en procesos como rosácea. En LES, IFD es insuficiente para realizar diagnóstico, se requiere correlación con hallazgos clínicos.
Fotoprovocación.
Por medio de técnicas estandarizadas, la exposición a radiación ultravioleta, se logra evidenciar la aparición de lesiones específicas de cada variedad de lupus. Las mismas aparecen con período de latencia de 4-8 días. A diferencia de otras fotodermatosis, como la erupción polimorfa solar, las lesiones aparecen más tempranamente, lo que sirve como diagnóstico específico y confirmatorio.
General.
Fiebre.
Cutáneo.
Alopecia no cicatrizal.
Úlceras orales.
LECSA-LED.
LECA.
Artritis.
Sinovitis en más de 2 articulaciones, dolor a la presión en más de 2 articulaciones o rigidez matinal por más de 30 minutos.
Neurología.
Delirio.
Psicosis.
Convulsiones.
Serositis.
Derrame pleural o pericárdico.
Hematología.
Anemia.
Trombocitopenia.
Hemólisis Autoinmune.
Renal.
Proteinuria.
Nefritis Lúpica.
Serológicos.
Antifosfolipídicos.
Hipocomplementemia. C3, C4.
Anti DNA.
Anti SM.
Parámetros de Laboratorio.
Hemograma. Anemia, leucopenia (linfopenia), trombocitopenia
Eritrosedimentación. Incrementa, hipogammaglobulinemia, en 20-50% de pacientes con LEC.
PCR. Puede incrementar en LEC/LES, puede indicar infección, pero también puede considerarse signo de serositis o artritis.
Creatininemia. Es muy poco sensible en los estadios tempranos de la nefritis lúpica. Sus niveles comienzan a incrementar con el compromiso avanzado del riñón. Estos niveles dependen de la edad del paciente y de su masa muscular. En estos casos son más confiables la filtración glomerular estimada (eGFR) o el clearence de ccreatinina en orina de 24hs
Sedimento urinario y proteinuria. Es requerido en la evaluación de la función renal.
Función hepática. Su alteración puede observarse en casos de síndrome de superposición con hepatitis autoinmune, o como evento adverso por fármacos.
CPK/LDH. Puede verse elevado CPK por miositis asociado a LES, aunque raramente, con evento adverso por hidroxicloroquina; la hemólisis puede generar un aumento de LDH.
Proteinograma. Hipoalbuminemia por nefritis lúpica, gammopatía monoclonal (2-4% de los casos).
ANA (Hep2). Es el principal screening para enfermedades del tejido conectivo. En LEC los títulos son habitualmente bajos (<1/320). Es condición necesaria para el diagnóstico de LES.
Ac Antifosfolípido. Más frecuentemente cardiolipinas, beta2glicoproteina, anticoagulante lúpico. Son criterio diagnóstico de LES. Son marcadores serológicos para síndrome de anticuerpo antifosfolipídicos. Pueden ser detectados en varias formas de LEC, pero infrecuentes, siendo más frecuentes en LES.
C3/C4. Criterio diagnóstico de LES. Es característico sus bajos niveles, siendo muy bueno para el monitoreo de la actividad de la enfermedad.
Diagnósticos Diferenciales..
Varios son los posibles diagnósticos comparables, pero el más importante es Erupción polimorfa solar, principalmente con las variantes LECSA y LECD. Pero un variado espectro de procesos pueden ser confundidos con alguna variante de LEC. En el caso de LECA: dermatomiosis, rosácea, dermatitis seborreica, tinea faciei (también en LECD), dermatitis perioral, eritema multiforme, necrolisis epidérmica tóxica; en LECSA los posibles son psoriasis, tinea corporis, erupción por drogas, eritema anular centrífugo, micosis fungoide, eritema multiforme, dermatitis seborreica. En LECD, queratosis actínica, sarcoidosis. En LEP, sarcoidosis, paniculitis, varias formas de linfoma, morfea, granuloma anular profundo, entre otros.
Protección Solar.
La estrecha relación entre LEC y su exacerbación por el sol, requiere una actitud dirigida a evitar la exposición, no sólo con el uso de protectores altos sino también con cambios de costumbres, como horarios de trabajo nocturnos, prendas de vestir adecuadas para repeler la luz, lentes con la protección adecuada, protectores solares con filtros aptos para UVA y UVB, aplicados 20-30’ previos a la exposición solar.
CLASI/RCLASI.
Confeccionado en 2005, Cutaneous Lupus Area and Severity Index, clasifica a los pacientes en leves (0-9), moderados (10-20) y severos (21-70). Una reducción del CLASI de 4 puntos se considera respuesta terapéutica efectiva. Una variante revisada (RCLASI), incluye en la evaluación signos como edema/infiltración o nódulos/pápulas profundos, lo que permite evaluar varias formas de LEC.
Calidad de Vida.
El compromiso cutáneo en LEC provoca frecuentemente alteraciones en la calidad de vida, debido a desfiguración, cicatrices, alopecia, lesiones mucosas, lesiones dolorosas.
No existe métodos específicos para evaluar calidad de vida en estos pacientes, aunque se recomienda el uso de DLQI o Skindex-29. El monitoreo del tratamiento debe considerar, siempre, los aspectos cosméticos, ya que influyen decididamente en la calidad de vida, principalmente las lesiones de la cara y el cuero cabelludo.
Pronóstico
Distintas variantes de LEC pueden, con diferentes frecuencias, iniciar el compromiso de órganos internos, como indicio de su viraje a LES. Entre las variantes de LECC, el LECD puede virar a LES en menos del 5%, siendo LECSA la variante que lo haga más frecuentemente, en 10-15% de los casos. Las manifestaciones extracutáneas más frecuentes son artralgias/artritis y proteinuria. Los pacientes con LECD diseminado tienen mayor compromiso extracutáneo y, por ende, mayor transformación en LES que los pacientes con formas localizadas.
El extenso compromiso cutáneo en LEC puede ser considerado como factor pronóstico.
Tratamiento
Los tratamientos clásicos utilizados son limitados y no han sido aprobados aún para LEC. La primera línea de tratamiento incluyen cambios en el estilo de vida como fotoprotección y dejar de fumar, junto con tratamientos tópicos como corticosteroides e inhibidores de la calcineurina. Las alternativas sistémicas son, con frecuencia, necesarias. Los antimaláricos de síntesis son, a menudo, utilizados como primera línea de tratamiento sistémico, pero también otros inmunomoduladores y supresores, como metotrexate, azatioprina, micofenolato sódico y mofetil, dapsona, talidomida y lenalidomide.
Las opciones biológicas tanto en las formas sistémicas como cutáneas son de gran interés. Las alternativas en este campo podemos diferenciarlas según el blanco hacia la cual están dirigidas: células B, células T o citoquinas. En la actualidad no existe ningún fármaco biológico aprobado para el tratamiento de LEC.
Rituximab
Anticuerpo monoclonal quimérico anti CD20, que induce la depleción de células B. Originalmente dirigido al tratamiento de linfomas B, actualmente se utiliza en varios procesos autoinmunes, como artritis Reumatoide, pénfigo vulgar y vasculitis ANCA +. Es muy escasa la información en relación a LEC. Muy pocos pacientes tratados con LEC (agudos, sub agudos o crónicos), pero todos han conseguido remisión parcial o total.
Belimumab
Anticuerpo monoclonal recombinante dirigido contra el factor estimulante de células B, que impide la unión con el receptor, por ende reduce la vida de los linfocitos B maduros. Actualmente es el único biológico aprobado para el uso en LES, en el caso de LEC sólo hay cohortes observacionales a partir de manifestaciones cutáneas de LES, por lo que no hay estudios firmes en LEC. En estudios con pacientes con LES con manifestaciones cutáneas han presentado reducción de SLEDAI y la dosis de corticoides requeridas al cabo de un año de tratamiento.
En otros estudios europeos en LES, se observó la reducción del CLASI en manifestaciones de ACLE y SCLE. En casos de LEC crónico se ha observado una gran mejoría de la manifestaciones en semana 16 medido por SLEDAI, PGA y CLASI. De todos modos se requieren más estudios controlados y de vida real para sacar conclusiones más serias.
Abatacept
Proteína de fusión de la molécula del antígeno 4 asociado al linfocito T citotóxico (CTLA 4) y a la porción Fc de la IgG1 la cual inhibe la activación de la Célula T coestimuladora. Aprobado para su uso en artritis reumatoide y artritis idiopática juvenil. Como consecuencia de estudios prospectivos y retrospectivos en LED se sugiere su acción sobre manifestaciones cutáneas de lupus inespecíficas, no observándose efectividad en LECC. Su acción sobre LECA y LECSA está aún evaluándose.
Lupuzor
Es un péptido sintético ligado a las proteínas de superficie de las células presentadoras de antígenos (HSPA8/HSC70). Actúa como inmunomodulador principalmente inhibiendo la reactividad de la Célula T. Los estudios preliminares hablan de la efectividad de Lupuzor en LE e indican un gran beneficio principalmente en las manifestaciones articulares y cutáneas a las 12 semanas, recibiendo 200microgrs/4 semanas.
Anifrolumab
Anticuerpo monoclonal anti IgG1k, totalmente humanizado, que liga al receptor de IFN-α/β/ω e interfiere en la señalización de todos los IFN tipo I. En los primeros estudios de LES de fase IIb, con procesos moderados a severos, consiguió una reducción del 50% en el CLASI a las 52 semanas y los resultados de seguridad en fase III son promisorios.
Sifalimumab
Anticuerpo monoclonal IgG1k, humanizado, que liga e interfiere con todas las variantes de IFN-α. En cuanto a los estudios preliminares están más avanzados que con Anifrolumab, mostrando una gran respuesta en estudios de fase II en pacientes con LES activo severo, así como en las manifestaciones cutáneas moderadas (CLASI >10). En semana 52 se ha observado una reducción de al menos 4 puntos en CLASI con todas las dosis probadas de 200-600 y 1200mg con un punto de máxima mejoría entre 20-24 semanas.
AMG811
Anticuerpo humanizado tipo IgG1 anti IFNγ. Actualmente sólo se cuenta con estudios fase I en pacientes con LED, concomitante o no con LES. No han mostrado diferencias significativas, respecto de placebo, en el CLASI.
Tocilizumab
Es un anticuerpo monoclonal IgG1 humanizado contra en receptor de la cadena α de la IL6, que inhibe la unión de la IL6 soluble y el componente de membrana. Ya aprobado para el uso en artritis reumatoide y artritis idiopática juvenil.
Estudios fase I y varios series de casos muestran su efectividad en LES, pero no hay estudios en LEC, aunque se observa mejoría de las manifestaciones cutáneas en el LES.
Sirukumab
Anticuerpo humano anti IL6. Sólo hay resultados de estudios fase I que no muestran con claridad su eficacia.
Terapias dirigidas contra IL: IL 12/23.
Ustekinumab
Es un anticuerpo monoclonal humano anti IgG1 kappa dirigido específicamente contra la unidad p40 de las IL12 y 23. Actualmente aprobado para su uso en psoriasis en placas y artritis psoriásica. Hay escasa estadística sobre su rol en LEC. Si se registra mayor información en LES con manifestaciones cutáneas observándose una mejoría de hasta el 50% en CLASI.
Bibliografía
Oshrat E Tayer-Shifman, Cheryl F. Rosen, Laura Wakani & Zahi Touma. Novel biological therapeutic approaches to cutaneous lupus erythematosus. https://doi.org/10.1080/14712598.2018.1513484.
Worm M, Zidane M, Eisert L and col. S2k guideline: Diagnosis and management of cutaneous lupus erythematosus – Part 1: Classification, diagnosis, prevention, activity scores. JDDG | 1610-0379/2021/1908.
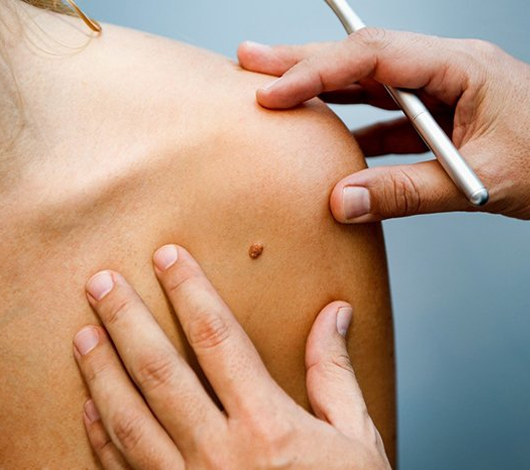
En investigación (Leer Más)

Nuestra Institución ha firmado un acuerdo con la Empresa Pharmexx, encargada de gerenciar ... (Leer Más)
El CANCER DE PIEL se clasifica de acuerdo a la célula originaria del mismo. (Leer Más)
Enfermedad inflamatoria crónica de la piel que produce lesiones escamosas engrosadas e inflamadas. (Leer Más)

Hiperhidrosis, Depilación Laser, Mesolifting/Mesoglow, Presoterapia secuencial... (Leer Más)

OSDE Binario, ACA Salud, ACCORD Salud, Unión Personal, IOMA, Poder Judicial. (Leer Más)

La luz ultravioleta forma parte del espectro de radiación de la luz solar, la que al descomponerse a través de un prisma. (Leer Más)
¿Es malo que los dedos me cambien de color con el frio? (Leer Más)

¿QUE ES LA CRIOCIRUGÍA? (Leer Más)